Cystic Fibrosis
Topic Overview
What is cystic fibrosis?
Cystic fibrosis is a genetic disease that causes mucus in the body to become thick and sticky. This glue-like mucus builds up and causes problems in many of the body’s organs, especially the lungs and the pancreas. People who have cystic fibrosis can have serious breathing problems and lung disease. They can also have problems with nutrition, digestion, and growth. The disease generally gets worse over time.
The life expectancy for people with cystic fibrosis has been steadily increasing.
What causes cystic fibrosis?
Cystic fibrosis is one of the most common genetic disorders in white children in the United States and Canada. It’s caused by a change, or mutation, in a gene. The changed gene is passed down in families. To pass on this disease, both parents must be carriers of the changed gene.
What are the symptoms?
Cystic fibrosis is usually diagnosed at an early age. The symptoms aren’t the same for everyone. But some common symptoms in a baby who has cystic fibrosis include:
- A blocked small intestine at birth. This prevents the baby from passing his or her first stool.
- Very salty sweat or skin.
- Diarrhea.
- Not growing or gaining weight the way that other children do.
- Breathing problems, lung infections, a cough that does not go away, and wheezing.
Other symptoms may also develop in childhood, such as:
- Clubbing (rounding and flattening) of the fingers.
- Rectal prolapse (when part of the rectum protrudes from the anus).
- Growths (polyps) in the nose or sinuses.
How is cystic fibrosis diagnosed?
Babies in the United States and Canada are tested for cystic fibrosis right after birth. Screening tests look for a certain health problem before any symptoms appear. The doctor may also notice the signs of cystic fibrosis during a routine exam.
If your child has a positive newborn screening test or symptoms of cystic fibrosis, your doctor will order a sweat test to see how much salt is in your child’s sweat. People with cystic fibrosis have sweat that is much saltier than normal. The doctor may also suggest a genetic test. Finding a high amount of salt in two sweat tests or finding certain changed genes will confirm a diagnosis.
How is it treated?
The types of treatment your child receives depends on what kinds of health problems the cystic fibrosis is causing and how your child’s body responds to different types of treatment. Doctors usually recommend a combination of medicines, home treatment methods (including respiratory and nutritional therapies), and other specialized care to manage the disease.
Health Tools
Health Tools help you make wise health decisions or take action to improve your health.
Cause
Cystic fibrosis occurs when a child inherits a changed (mutated) gene from both parents. The changed gene causes problems with the way salt and water move in and out of the cells that make mucus, causing the mucus to be thick and sticky. This kind of mucus makes it hard for the body to keep certain organs clean and healthy.
Many people don’t know they have the changed gene. If you are the parent of a child who has cystic fibrosis, it is important to remember that nothing you did caused the disease.
Symptoms
Symptoms of cystic fibrosis are usually caused by the production of thick, sticky mucus throughout the body. Symptoms vary from person to person. They aren’t always obvious in childhood.
Early symptoms
Symptoms of cystic fibrosis in a baby or young child may include:
- A blocked small intestine at birth.
- Salty sweat or skin.
- Wanting to eat more or less than normal, having little energy, or losing weight.
- Unusual bowel movements.
- The child may have diarrhea that doesn’t go away, large and greasy stools, very smelly stools, or constipation.
- If the intestines become blocked, the child’s belly may stick out. The child may not be able to have a bowel movement.
- Breathing problems or getting tired easily while playing.
- A cough that doesn’t go away or wheezing.
Later symptoms
Over time, symptoms may get worse and cause problems such as:
- Coughing up mucus that sometimes has blood in it.
- Difficulty exercising or not being able to exercise.
- Rectal prolapse, which means that part of the rectum protrudes from the anus.
Other symptoms
More symptoms may develop during late childhood or early adulthood. They include:
- Clubbing (rounding and flattening) of the fingers.
- Growths (polyps) in the nose or sinuses.
- Not being able to have children (infertility).
What Happens
Although cystic fibrosis generally follows certain patterns, each person’s symptoms depend on what is happening with his or her mucus-producing cells. These kinds of cells are found throughout the body in many different organs and systems, including the:
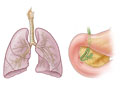
- Lungs and respiratory system. People with cystic fibrosis have thick and sticky mucus that traps bacteria. This causes lots of lung infections and often causes permanent lung damage. Bronchiectasis, which is caused by long-lasting airway inflammation, is common.
- Pancreas and digestive system. Mucus can interfere with how the pancreas works. This can make it hard for the child to absorb nutrients from food.
- Sweat glands. You may notice that your child has unusually salty skin. Cystic fibrosis can cause a person to become easily dehydrated or to have very low salt levels.
- Reproductive organs. Almost all men who have cystic fibrosis are unable to father a child. Women who have cystic fibrosis may have more difficulty getting pregnant than other women.
- Skeletal system. People who have cystic fibrosis may have weaker bones than other people. This is because their bones contain lower levels of minerals. Weakened bones can lead to bone fractures or osteoporosis. Cystic fibrosis can also cause swollen or painful joints (arthropathy or arthritis). These problems are more common in adults than in children.
Life expectancy
The life expectancy for people with cystic fibrosis has been steadily increasing. On average, people who have cystic fibrosis live into their mid-to-late 30s. But new treatments are helping some people to live into their 40s and longer. People who have a mild form may have a normal life expectancy.
Even though cystic fibrosis can’t be cured, the treatments continue to get better. And there are new treatments that target the cystic fibrosis gene defect.
What Increases Your Risk
Cystic fibrosis is a genetic disorder. It is an autosomal recessive disease. This means that to get the disease, you must inherit the changed (mutated) gene from both parents. Keep in mind:
- If a person inherits the changed gene from only one parent, he or she will not get cystic fibrosis but will be a carrier of the disease. Carriers may pass the gene defect on to their children.
- If you are planning a pregnancy and you are white, talk to your doctor about having a genetic test to find out your chances of having a child with cystic fibrosis. The disease is much more common in non-Hispanic white people than in people of other races and ethnic backgrounds.
When should you call your doctor?
It is important to diagnose and treat cystic fibrosis early. Call your doctor if your child:
- Often has lung infections (pneumonia), colds, a cough, shortness of breath, or wheezing.
- Coughs up mucus that contains blood.
- Doesn’t gain or stay at the same weight.
- Has smelly, large, greasy stools or diarrhea.
- Tires easily during activity.
- Has rounded, flat fingertips (clubbing).
Call your doctor if your child who has been diagnosed with cystic fibrosis gets worse in any way. Typically, this is when your child:
- Has increased coughing or has a cough that is getting worse.
- Has new wheezing or has wheezing that is getting worse.
- Has more trouble breathing than usual.
- Has lost weight or is not gaining weight, for no clear reason.
- Is having symptoms that you and your doctor have discussed as being more serious problems.
Watchful waiting
Watchful waiting is a wait-and-see approach. It’s not a good idea for people who have cystic fibrosis. If your child has any signs of cystic fibrosis, even if they seem to be mild, call the doctor right away.
Who to see
The following doctors can diagnose and treat cystic fibrosis:
Other health professionals may also be involved in your child’s care:
- Respiratory therapist
- Physical therapist
- Registered dietitian
- Exercise physiologist
- Social worker
- Psychologist
- Pharmacist
Cystic fibrosis care centers offer the best, most comprehensive treatment available by addressing medical, nutritional, and emotional needs. You can find one by contacting the Cystic Fibrosis Foundation at www.cff.org.
Exams and Tests
Tests can help find out if a person has cystic fibrosis or has the changed gene that can be passed on to a child. Adults may want to get tested during a pregnancy or when they are planning for a pregnancy. People can be tested at any age to see if they have cystic fibrosis.
Diagnosis
A medical history and a physical exam are often the first steps in diagnosing cystic fibrosis, followed by screening or lab tests.
The diagnosis of cystic fibrosis requires one of the following:
- Your child has early symptoms. These may include diarrhea that does not go away; large, greasy or very smelly stools; constipation; not wanting to eat; or losing weight. To learn more, see Symptoms.
- A brother or sister who has cystic fibrosis.
- A positive newborn screening test.
Also, there must be at least one of the following:
- A genetic test showing that a person inherited one or two defective cystic fibrosis transmembrane regulator (CFTR) genes. This testing can be done using blood or a sample from the mother’s womb before birth (chorionic villus sampling or amniocentesis).
- Two positive sweat tests on different dates. Sweat tests measure the level of salt in sweat. People with cystic fibrosis have more than the normal amount of salt in their sweat. If a genetic test detects two defective CFTR genes, then just one sweat test result may be all that is needed to confirm a diagnosis.
- An abnormal nasal potential difference test. This test uses electrodes on the lining of the nose to see how well salts flow into and out of cells.
Monitoring cystic fibrosis
Certain tests can help your doctor monitor your child’s cystic fibrosis. These tests include:
- Lung function tests to find out how healthy the lungs are by checking how well air moves into and out of the lungs.
- A throat culture or sputum culture to see what kinds of bacteria are causing any infections your child may have.
- A chest X-ray to take a picture of the chest, including the heart and lungs.
- A CT scan to find any serious disease in the lungs, pancreas, or other organs.
- Blood tests, such as the oral glucose tolerance test and liver function. These tests are to see if there are any complications of cystic fibrosis.
- A stool analysis to see how well your child is absorbing and digesting fat and other nutrients.
- An arterial blood gas analysis to measure the levels of oxygen and carbon dioxide in the blood. This test shows how well the lungs are working.
Early detection
Both newborns and adults can be tested for the changed (mutated) gene that causes cystic fibrosis. These tests include:
- Newborn screening. Levels of a type of digestive enzyme are measured from a blood sample. High levels of this enzyme suggest cystic fibrosis. Some newborns may also have a genetic test.
- Genetic test for adults. These tests identify the most common defects in the CFTR gene. Genetic testing can be done during pregnancy through chorionic villus sampling or amniocentesis. The test can also be done before pregnancy, to help couples find out if either or both of them carry a defective CFTR gene.
- If both parents carry the changed gene, there is a 25% (1-in-4) chance that their child will have no genetic problem. There is a 25% chance that their child will have cystic fibrosis. And there is a 50% (1-in-2) chance that their child will be a carrier.
- If only one parent is a carrier of the changed gene, the child will not have cystic fibrosis. But there is a 50% chance that the child will be a carrier.
If you are interested in a genetic test for cystic fibrosis, talk with your doctor about the test. Genetic counseling can help you understand your test results. Genetic testing may involve certain ethical, legal, and religious issues.
Treatment Overview
After a child is diagnosed, a team of health professionals will build a treatment plan based on the child’s specific health problems. Following a treatment plan will help your child live a longer, healthier life.
Your child will likely have ongoing respiratory therapy, digestive therapy, and treatment with medicines such as antibiotics. Regular medical care, home treatment such as postural drainage, and taking steps to reduce infection can help people with cystic fibrosis lead relatively normal lives.
The best treatment available is generally found at cystic fibrosis care centers. These centers address the medical, nutritional, and emotional needs. You can find one by contacting the Cystic Fibrosis Foundation at www.cff.org.
Doctor visits and immunizations
Regular visits with the team of health professionals involved in your child’s care are important. Your doctor will want to make sure that your child is eating properly and is gaining weight and growing at a normal rate. The doctor will record your child’s weight, height, and head size in order to keep track of how your child is developing over time.
Lab tests can help your doctor know how serious the disease is and how it is affecting your child’s body.
Your doctor will ask you about your child’s immunizations and will schedule any shots that are needed. Children with cystic fibrosis should have all the recommended shots, plus pneumococcal shots. To learn more, see the topic Immunizations.
Respiratory therapy
Respiratory therapy is any treatment that slows down lung damage and improves breathing. The focus of this therapy is on reducing infection and getting rid of mucus to keep the lungs healthy. Medicines may be used to control the amount and thickness of mucus.
Other ways to help remove mucus from the lungs involve certain types of movements, coughing, or exercises known as airway clearance techniques.
People with severe lung disease may need to use oxygen at home.
Digestive therapy
This treatment works to replace certain digestive enzymes, to make sure the body absorbs all the vitamins and minerals it needs, and to prevent or treat intestinal blockages. Digestive therapy involves:
- Digestive enzyme replacement therapy (such as with Creon or Pancreaze), to help the intestines absorb nutrients from food.
- Nutritional therapy to help replace lost nutrients. This may include taking vitamins; eating high-calorie, high-fat foods; drinking nutritional drinks; getting fed through a tube in the stomach; and, in some cases, receiving intravenous nutrient supplementation.
- Preventing intestinal blockages with stool softeners (to avoid constipation) and enemas.
Treating complications
Serious cystic fibrosis problems or complications occur when the respiratory system or digestive system becomes damaged. Most people who have complications will need to stay in the hospital. Treatment for complications may include medicines or surgery, depending on the person’s age and symptoms.
The doctor may do tests, such as a chest X-ray, to know what kinds of problems your child is having.
Other treatments for complications from cystic fibrosis may include:
- Blood transfusions and medicines to treat the bleeding (embolization therapy), if your child is coughing up large amounts of blood. Coughing up small amounts of blood is normal for people who have cystic fibrosis. But coughing up large amounts of blood can be life-threatening.
- Placement of a semipermanent intravenous (IV) tube to give your child antibiotics frequently without having to place a line in the vein each time.
Home care for cystic fibrosis
Home treatment is very important. It can make a person with cystic fibrosis feel better and live longer. Here are some things you can do at home, or help your child do, to help prevent more serious health problems like lung infections:
- Don’t smoke. And avoid secondhand smoke.
- Use airway clearance techniques, such as postural drainage and chest percussion.
- Eat nutritious, high-calorie foods.
- Exercise.
- Drink plenty of fluids.
- Add salt to foods, especially during hot weather.
- Get all recommended vaccines and practice good hygiene. Also keep clean any breathing equipment you use for your treatment.
As children with cystic fibrosis get older, it is important for them to learn how to help care for themselves. Even though it can be hard to follow a treatment plan every day, there are many benefits of home treatments. Skipping a treatment may not make a person feel worse right away. But it raises the chances of having more serious problems later.
Getting support
Handling the challenges of caring for a child who has cystic fibrosis can be difficult. Take good care of yourself, physically and emotionally, so that you can give your child the best care possible.
Many people with cystic fibrosis and their families need emotional support to help them live with this genetic disease. Support groups, counseling, and education about the disease can be very helpful not only for people who have cystic fibrosis but also for their families.
It is also important to talk about the kind of medical procedures you want or don’t want for yourself or for your child.
Research for new treatments
Medical researchers are looking at gene transfer therapy. It involves introducing healthy genes into the lung cells of people who have cystic fibrosis.
Researchers are also investigating protein repair therapy, or protein assist therapy. This treatment involves taking medicines that help the defective protein work more normally to allow a small amount of salt and water to move out of cells.
Gene transfer and protein repair therapies are in the experimental, developmental stages. Talk to your doctor about clinical trials for these and other new treatments being studied.
Prevention
Cystic fibrosis is a genetic disorder that cannot be prevented. But there are many things you can do to help your child live a happy and healthy life. To learn more, see the Treatment Overview.
Medications
Medicines for cystic fibrosis help keep the lungs as healthy as possible, reduce and control mucus in the lungs, and replace digestive enzymes.
Medicine choices
Medicines to treat infections
- Antibiotics (such as ciprofloxacin and tobramycin)
Medicines to open airways in the lungs or keep them open
- Bronchodilators (such as albuterol or salmeterol), which are used to make breathing easier. They may also make it easier to cough up mucus.
- Anticholinergics (such as Atrovent)
Medicines to control the amount and thickness of mucus
- DNase (such as Pulmozyme). It is used to thin mucus in the lungs.
- Mucolytics (such as acetylcysteine), to thin mucus in the lungs and the intestines. These aren’t used very much, though, because they can irritate the lungs.
- Saltwater solution (hypertonic saline). This is sometimes used to help clear mucus from the lungs. It is low-cost, and it may help reduce inflammation in the airways.footnote 1, footnote 2
Medicines to reduce inflammation
- Nonsteroidal anti-inflammatory drugs (NSAIDs) (such as ibuprofen)
- Membrane stabilizers (such as cromolyn)
- Corticosteroids (such as fluticasone or prednisone)
Medicines to replace the effect of digestive enzymes
- Enzyme replacement therapy (such as Creon or Pancreaze)
Medicines to correct mutated genes
- Medicines such as ivacaftor (Kalydeco), lumacaftor/ivacaftor (Orkambi), and tezacaftor/ivacaftor (Symdeko) can correct some types of gene mutations that cause cystic fibrosis.
These medicines cost a lot, but there may be programs to help pay for them.
Talk to your doctor if you want to know more.
What to think about
Some medicines work better for some people than for others. A medicine that works well for one person may not work for someone else. It can take time to find the medicines that work best for you or your child.
Surgery
Surgery may be needed to treat complications of cystic fibrosis. Procedures may include:
- Lung transplant, for people who have severe lung disease.
- Chest tube drainage and possible thoracoscopy, to treat a collapsed lung (pneumothorax).
- Repair of an intestine that has collapsed inside itself or removal of a bowel obstruction.
- Removal of nasal polyps or endoscopic sinus surgery.
If your child isn’t able to get all the nutrients he or she needs from food, a feeding tube may be placed in your child’s stomach.
What to think about
Lung transplant surgery is generally recommended only for people who have severe lung damage, because the risks can be greater than the benefits. Not everyone is a good candidate for an organ transplant. If tests show that you are a good candidate, you are put on a waiting list. You may have to wait days, months, or years for your transplant. Be patient, and ask your doctor what you can do while you’re waiting. Guidelines from the United Network for Organ Sharing (UNOS) are intended to shorten the wait time for donor lungs.
For more information on transplants, see the topic Organ Transplant.
References
Citations
- Elkins MR, et al. (2006). A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. New England Journal of Medicine, 354(3): 229–240.
- Wark P, McDonald VM. (2009). Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews (2).
Other Works Consulted
- ACOG Committee on Genetics (2002, reaffirmed 2006). Genetics and molecular testing. ACOG Technology Assessment in Obstetrics and Gynecology, 100(1): 193–211.
- American Academy of Pediatrics (2009). Nutrition in cystic fibrosis. In RE Kleinman, ed., Pediatric Nutrition Handbook, 6th ed., pp. 1001–1020. Elk Grove Village, IL: American Academy of Pediatrics.
- American Academy of Pediatrics (2014). Nutrition in cystic fibrosis. In Pediatric Nutrition, 7th ed., pp. 1113–1145 . Elk Grove Village, IL: American Academy of Pediatrics.
- Balfour-Lynn IM, Welch K (2012). Inhaled corticosteroids for cystic fibrosis. Cochrane Database of Systematic Reviews (11).
- Borowitz D, et al. (2009). Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. Journal of Pediatrics, 155(6): S73–S93.
- Boucher RC, et al. (2010). Cystic fibrosis. In R Mason et al., eds., Murray and Nadel’s Textbook of Respiratory Medicine, 5th ed., vol. 1, pp. 985–1022. Philadelphia: Saunders.
- Dovey ME (2006). Cystic fibrosis. In FD Burg et al., eds., Current Pediatric Therapy, 18th ed., pp. 457–461. Philadelphia: Saunders Elsevier.
- Elkins MR, et al. (2006). A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. New England Journal of Medicine, 354(3): 229–240.
- Ernst MM, et al. (2011). Developmental and psychosocial issues in cystic fibrosis. Pediatric Clinics of North America, 58(4): 865–885.
- Farrell PM, et al. (2008). Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. Journal of Pediatrics, 153(2): S4–S14.
- Flume PA, et al. (2009). Cystic fibrosis pulmonary guidelines: Airway clearance therapies. Respiratory Care, 54(4): 522–537.
- Flume PA, et al. (2009). Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. American Journal of Respiratory and Critical Care Medicine, 180(9): 802–808.
- Grosse SD, et al. (2004). Newborn screening for cystic fibrosis: Evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR, 53(RR-13): 1–36.
- Gustafsson PM, et al. (2008). Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax, 63(2): 129–134.
- Jones AP, Wallis C (2010). Dornase alfa for cystic fibrosis. Cochrane Database of Systematic Reviews (3).
- Nash EF, et al. (2009). Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database of Systematic Reviews (1).
- Organ Procurement and Transplantation Network (2013). Organ distribution: Allocation of thoracic organs, Policy 3.7. Available online: http://optn.transplant.hrsa.gov/policiesAndBylaws/policies.asp.
- Ratjen F, McColley S (2012). Update in cystic fibrosis 2011. American Journal of Respiratory and Critical Care Medicine, 185(9): 933–936.
- Southern KW, et al. (2009). Newborn screening for cystic fibrosis. Cochrane Database of Systematic Reviews (1).
- Wark P, McDonald VM. (2009). Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews (2).
Current as of: December 12, 2018
Author: Healthwise Staff
Medical Review:John Pope MD – Pediatrics & Kathleen Romito MD – Family Medicine & R. Steven Tharratt MD, MPVM, FACP, FCCP – Pulmonology, Critical Care Medicine, Medical Toxicology



This information does not replace the advice of a doctor. Healthwise, Incorporated, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use. Learn how we develop our content.